
Multi-omics Analysis Service
Online Inquiry- Service Details
- Case Study
The process of occurrence and regulation of life phenomena is extremely complex. The same biological process may occur in different subcellular locations, and the genes involved may be regulated by different regulatory mechanisms. In the process of the occurrence and development of complex diseases such as tumor, autoimmune disease and metabolic disease, as well as in the life phenomena such as stem cell differentiation, embryonic development and species evolution, multilevel changes and regulation such as genome, transcriptome, proteome and epigenetic inheritance may be involved.
Multi-group analysis can obtain a large amount of data in a short time. The research results can be obtained quickly by combining with the key analysis. The data of omics at different levels can be mutually verified, making the end of the experiment obtained by comprehensive analysis more reliable. The multi-layer omics integration can overcome the defects caused by single-layer omics in detecting the difference of preference. The multilayer omics integration can make up for the defect caused by the difference of preference in single group detection. There is a tendency to magnify the effect from gene, protein to metabolism, and it is easier to find the difference by analyzing the downstream omics data.
As a good partner of pharmaceutical companies and research institutions, Creative Proteomics can provide genomics, transcriptomics, proteomics and metabolomics experiments and data analysis services, as well as provide multi-omics joint analysis to accelerate your project progress.
Advantages of Subcellular Multi-omics Analysis Service
- In-depth exploration of candidate pathogenic factors: integrate genomics and transcriptomics data, and analyze the multi-layer response of disease occurrence. These include changes in expression levels caused by gene mutations, as well as various forms of isomers and feedback regulation in transcriptional regulation, translation, and post-translational regulation. Target pathogenic targets based on changes in candidate factors at different levels.
- Construct gene regulatory network: The interaction between genes, mRNA, regulatory factors, and proteins in an organism forms a network relationship. Through the construction of gene regulatory networks, the various molecules are connected to each other. It will help to clarify the regulation and causality between the various molecules, and promote the research on the molecular mechanism and genetic basis of complex traits in genetic diseases.
- Multi-omics joint analysis: For example, the impact of candidate genes screened at the DNA level at the transcription level, mutation analysis at the RNA level for genes that are significantly differentially expressed at the RNA level, and integrated analysis based on metabolite related enzymes and differential genes.
We Provide the Following Services, including but not Limited to:
Quantitative Proteomics and Metabolomics: Measures changes in protein and metabolite abundance, facilitating precise comparisons between different conditions or time points. Investigates the proteome and metabolome of specific subcellular compartments, unveiling compartment-specific molecular processes and functions.
Proteomics and Transcriptomics: Integrating transcriptomics (RNA-Seq) and proteomics data allows you to correlate gene expression with protein abundance, helping to understand how changes at the transcriptional level relate to changes in the proteome. For example, if you want to investigate post-transcriptional regulation of gene expression.
Metabolomics and Transcriptomics: Analyzing both metabolites and gene expression data can uncover how metabolic processes are regulated at the transcriptional level. It's useful for understanding the regulatory mechanisms of metabolic pathways.
Proteogenomics: Integrates proteomics and genomics data to identify novel protein-coding genes, mutations, and fusion proteins relevant to disease and biomarker discovery.
Metagenomics and Metaproteomics: Analyzes metagenome and metaproteome to explore microbial community functions and discover biomarkers in environmental and microbiome studies.
Pharmacoproteomics and Metabolomics: Evaluates the effects of drugs on the proteome and metabolome, supporting drug development.
Single-Cell Proteomics and Metabolomics: Investigates the molecular composition of individual cells, revealing cellular heterogeneity.
Data Integration and Bioinformatics: Performs data integration, statistical analysis, and interpretation to extract meaningful insights from multi-omics datasets.
Multi-omics Analytical Techniques
Liquid Chromatography-Mass Spectrometry (LC-MS): LC-MS is widely used for proteomics. Instruments like the Thermo Fisher Q Exactive, Waters Xevo G2-XS QTof, or Agilent 6550 iFunnel are commonly employed for protein identification and quantification.
Selected Reaction Monitoring (SRM): Applied on triple quadrupole mass spectrometers, such as the AB SCIEX QTRAP 6500, for targeted quantification of specific proteins.
Data-Independent Acquisition (DIA): Instruments like the SCIEX TripleTOF series are used for comprehensive proteome analysis.
Gas Chromatography-Mass Spectrometry (GC-MS): GC-MS instruments like the Agilent 7890B/5977A are used for the analysis of small molecules and volatile compounds.
Liquid Chromatography-Mass Spectrometry (LC-MS): Similar to proteomics, LC-MS is also employed for metabolomic analysis.
Genomics:
Genomics focuses on the complete DNA sequence of an organism. High-throughput DNA sequencing technologies like Illumina's Next-Generation Sequencing (NGS) platforms, PacBio's Single Molecule Real-Time (SMRT) sequencing, and Oxford Nanopore's nanopore sequencing are commonly used. Mass spectrometry is not a primary tool in genomics.
Transcriptomics:
Transcriptomics studies the entire set of RNA transcripts in a cell or tissue. Illumina's NGS platforms, as well as other RNA-Seq methods, like those offered by PacBio, are widely utilized.
Multi-Omics Data Analysis
| Data Type | Data Processing | Key Analysis Steps |
|---|---|---|
| Genomics | Sequencing (NGS, Sanger) | 1. Quality Control of Sequencing Data |
| 2. Read Mapping to a Reference Genome | ||
| 3. Variant Calling (SNPs, Indels, etc.) | ||
| 4. Structural Variant Detection (optional) | ||
| Transcriptomics | RNA-Seq | 1. Quality Control of RNA-Seq Data |
| 2. Alignment and Quantification of Reads | ||
| 3. Differential Expression Analysis | ||
| 4. Pathway Analysis | ||
| Proteomics | Mass Spectrometry | 1. Data Preprocessing (e.g., noise removal) |
| 2. Peptide Identification and Quantification | ||
| 3. Protein Inference and Quantification | ||
| 4. Differential Expression Analysis | ||
| Metabolomics | Mass Spectrometry | 1. Data Preprocessing (e.g., noise removal) |
| 2. Metabolite Identification and Quantification | ||
| 3. Metabolite Annotation (e.g., in databases) | ||
| 4. Differential Analysis of Metabolites | ||
| Epigenomics | Bisulfite Sequencing | 1. Quality Control of Sequencing Data |
| 2. DNA Methylation Analysis | ||
| Histone Modification Analysis | 1. ChIP-Seq Data Quality Control | |
| 2. Peak Calling and Annotation | ||
| Integrative Analysis | Multi-Omics Data | 1. Data Integration and Normalization |
| 2. Correlation and Network Analysis | ||
| 3. Pathway Enrichment Analysis | ||
| 4. Integration with Clinical Data (if applicable) |
In addition to the above integrated analysis, Creative Proteomics can customize exclusive solutions for you based on your projects and needs. If you want to know more about the analysis service of multi-omics, please contact us.
Case: Differential Expression and Clinical Significance of NUF2 in Non-Small Cell Lung Cancer
Background
Non-Small Cell Lung Cancer (NSCLC) is a major cause of cancer-related deaths globally. Identifying key genes associated with NSCLC and understanding their molecular mechanisms is crucial for improving diagnosis, treatment, and prognosis. NUF2, a gene linked to cell cycle regulation, has been implicated in various cancers, but its role in NSCLC is not fully understood.
Samples
The study utilized a dataset comprising 1,144 NSCLC patient samples, categorized into LUAD (Lung Adenocarcinoma) and LUSC (Lung Squamous Cell Carcinoma) subtypes, obtained from TCGA and GEO databases. Normal tissue samples served as controls.
Technical Methods
Data Collection and Processing: The study collected data from TCGA and GEO databases, and a flow diagram illustrated the data acquisition process.
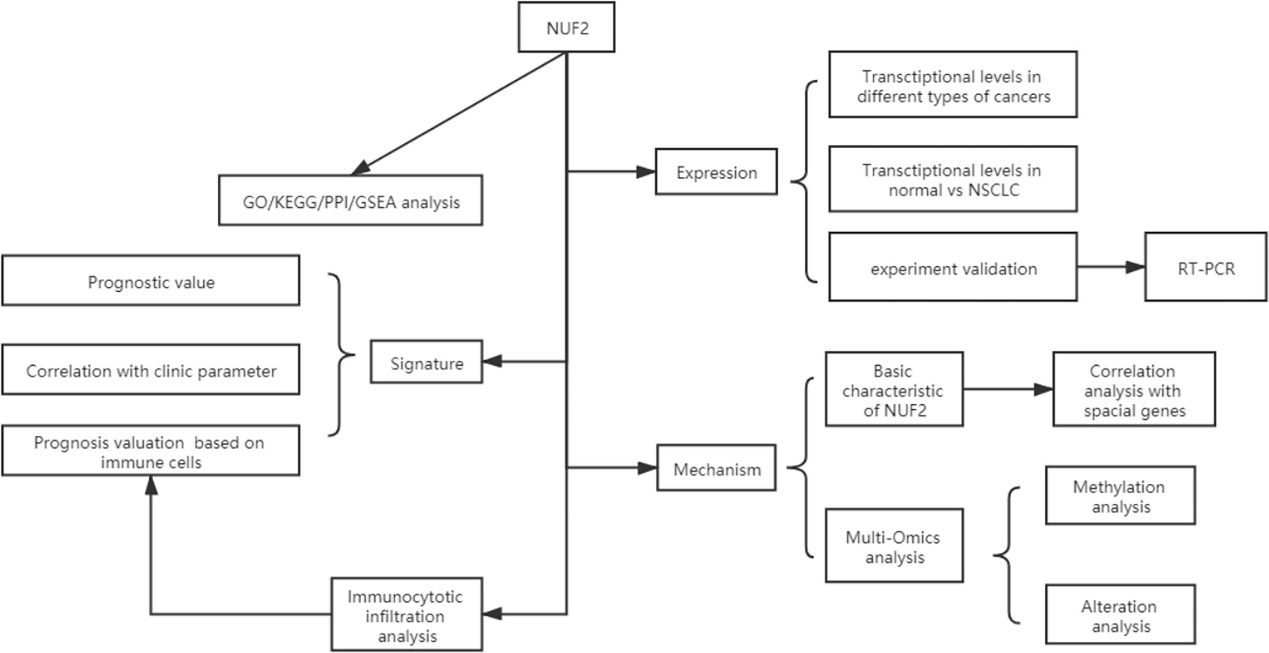 Flow diagram of data collection and method implementation in this work.
Flow diagram of data collection and method implementation in this work.
NUF2 Expression Analysis:
- Utilized Oncomine and TIMER databases to compare NUF2 mRNA expression in NSCLC vs. normal tissue.
- Examined NUF2 expression in LUAD (GSE32863) and LUSC (TCGA-FPKM).
- Conducted a differential expression analysis using GSE77803 data and visualized the results in a volcano plot.
Prognosis and Clinical Features Analysis:
- Investigated the association between NUF2 expression and patient prognosis, dividing them into high and low expression groups based on median NUF2 levels.
- Analyzed NUF2 expression in relation to clinical factors such as cancer stage and smoking habits.
Functional Annotation and Protein-Protein Interaction (PPI):
- Enrichment analysis revealed NUF2's involvement in organelle fission, nuclear division, and spindle-related processes.
- Explored the Protein-Protein Interaction network of NUF2 and identified key genes associated with NUF2.
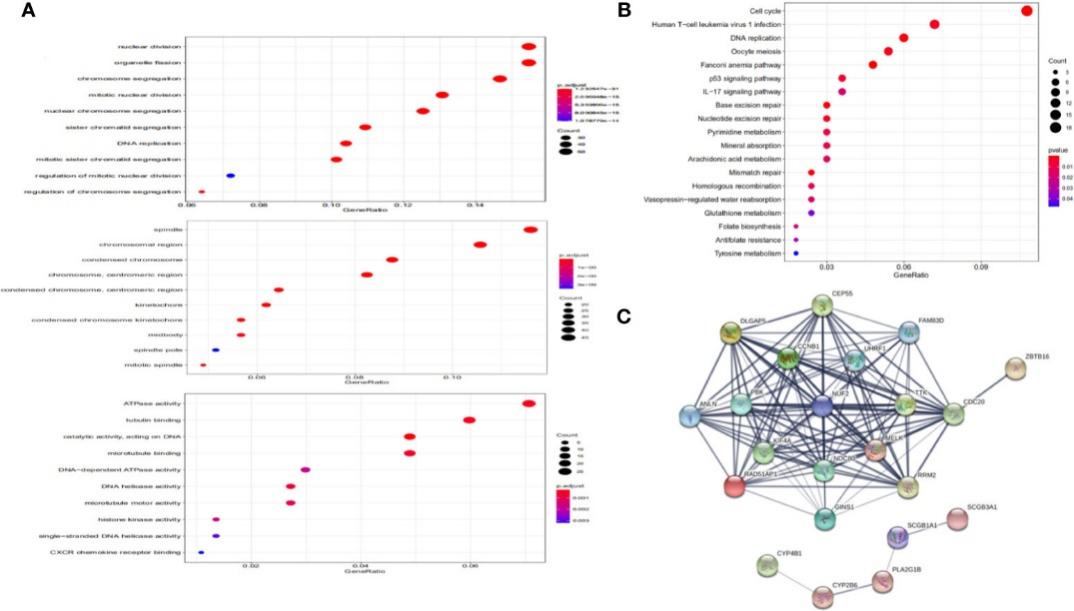 Functional annotation and protein-protein interaction.
Functional annotation and protein-protein interaction.
NUF2 DNA Methylation and Mutation Analysis:
- Investigated the DNA methylation status of NUF2 in LUAD and LUSC, finding differential methylation patterns.
- Explored NUF2 alterations and their frequency using whole-exome sequencing data, indicating NUF2 amplification as a common alteration.
Immune Correlation Analysis:
- Investigated the relationship between NUF2 expression and immune cell markers.
- Examined the association between NUF2 expression and immune cell infiltration in LUAD and LUSC.
Prognosis Analysis Based on Immune Infiltration:
- Analyzed how NUF2 expression impacted patient prognosis in the context of immune cell infiltration.
- Investigated the influence of NUF2 on different immune cell subtypes in LUAD and LUSC.
Results
- NUF2 expression was significantly higher in both LUAD and LUSC compared to normal lung tissue.
- Higher NUF2 expression was associated with poorer overall survival in LUAD, suggesting its prognostic potential.
- Functional annotation revealed NUF2's enrichment in cell cycle-related processes.
- NUF2 displayed differential DNA methylation patterns between LUAD and LUSC.
- NUF2 alterations, primarily gene amplifications, were common in NSCLC, with a potential impact on prognosis. Additionally, NUF2 was negatively correlated with immune cell markers and influenced immune cell infiltration in NSCLC, with potential implications for immunotherapy response.
Reference
- Chen, Mengqing, et al. "Integrative multi-omics analysis of identified NUF2 as a candidate oncogene correlates with poor prognosis and immune infiltration in non-small cell lung cancer." Frontiers in Oncology 11 (2021): 656509.
* For Research Use Only. Not for use in diagnostic procedures.
