
X-Ray Crystallography (XRC) Service
Online InquiryX-ray diffraction is an important method to determine the crystal structure of proteins. There is currently no tool that can directly observe the arrangement of atoms and groups inside a protein. Although the electron microscope is close to seeing the outline of macromolecules, it is limited to revealing the size, shape, symmetry, and aggregation state of the molecules. The X-ray crystallography method can be used to indirectly study the spatial structure of protein crystals. The study of crystal structure helps to understand matter at the atomic level.
Creative Proteomics has professional crystal diffraction equipment and rich experience in protein structure analysis, and can provide one-stop X-ray crystallography service, which aims to provide you with scientific support for protein crystal structure analysis.
The Process of X-Ray Crystallography (XRC) Service
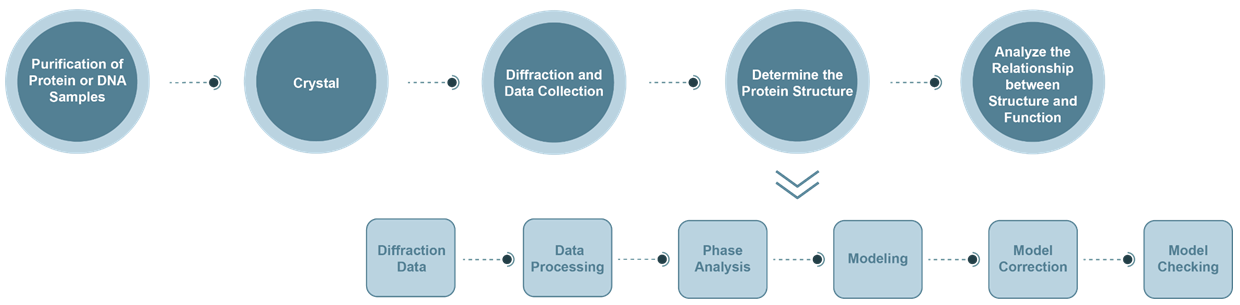
- Diffraction data collection: The wavelength of X-rays generated by synchrotron radiation can be varied. It has the advantages of higher light source intensity and better resolution.
- Data analysis: By analyzing the recorded diffraction data, the crystal system to which the crystal belongs and the corresponding Bravi lattice, as well as the miller index and corresponding intensity of each diffraction point in reciprocal space can be obtained. It mainly includes: a. indexing; b. intensity integration, merger, reduction of amplitude; c. evaluation of the quality of protein crystal diffraction intensity data.
- Crystal structure analysis: Crystal diffraction is actually the superposition of X-ray diffraction by the electron density of each atom in the crystal. The diffraction data reflects the result of Fourier transform of the electron density, which is expressed by the structure factor. The inverse Fourier transform of the structural factors is used to obtain the electron density distribution in the crystal.
- Phase analysis:
a. Molecular replacement method: If the molecular structure of a homologous protein of a protein to be measured is known, we will choose the molecular replacement method to solve its diffraction phase problem. The advantage of this method is that it is not necessary to prepare heavy atom derivatives, as long as a set of diffraction data is collected. Applicable conditions: The target protein A has a homologous protein structure B with a homology of more than 30%.
b. Multiple Isomorphic Replacement (MIR): Use heavy metal atoms with large X-ray scattering capability as the identification atoms. Such a macromolecule substituted with a heavy atom should have the same unit cell parameters and space group as the original crystal without a heavy atom, and most of the atoms have the same position. From the diffraction data of these heavy atom-containing crystals, a method based on the Paterson method can be used to solve the position of the heavy atom and calculate its structural factor and phase angle based on this. The phase angle relationship is then used to calculate the phase angle of the original crystal without heavy atoms, thereby solving the structure.
c. Multiwavelength Anomalous Scattering (MAD): This method is often used in combination with the heavy atom isomorph substitution method. After receiving the diffraction data of the isomorphic substitution, the wavelength of the incident ray is changed to the absorption edge of the heavy atom, and the data is collected again. This set of data has anomalous scattering. The two sets of data can be used to find the phase.
Technology Platform
Bruker AVANCEIII HD 800MHz and Bruker AVANCE III HD 600MHz two liquid nuclear magnetic resonance spectrometers are available
Advantages of X-ray Crystallography Service:
- Provides rich and unique information about crystalline materials
- Ability to uniquely identify specific crystal phases
- Can measure lattice spacing
- Can analyze complete wafers up to 300mm
- Low or no sample preparation requirements
- Non-destructive, use environmental conditions for all analyses
Want to Know about Other Subcellular Protein Structure Analysis Techniques?
References
- Drenth J. Principles of protein X-ray crystallography. Springer Science & Business Media, 2007.
- Smart O S, Horský V, et al. Validation of ligands in macromolecular structures determined by X-ray crystallography. Acta Crystallographica Section D: Structural Biology, 2018, 74(3): 228-236.
* For Research Use Only. Not for use in diagnostic procedures.
